Revision of the EU Clinical Trial Provision
On 16 April 2014 the Clinical Trials Regulation EU No 536/2014 [1] was adopted and will repeal the Clinical Trials Directive 2001/20/EC [2]. Once applicable, the Regulation 536/2014 will automatically be binding for all member state countries and will replace the (differing) national legislations on clinical trials in order to achieve “a greater level of harmonisation of the rules for conducting clinical trials throughout the EU.” [3] The processes of a harmonised approach to submission, assessment and report of clinical trails “are to be supported by a EU portal and EU database which will ensure a centralised workflow with monitoring by the relevant parties.” [4] The set-up of the EU portal and database has become the determining factor for the date of entry into application of the Clinical Trials Regulation. While the portal’s go live date had previously been planned for 2018, it now is postponed to 2020 due to technical reasons. [5] In consequence of the revision of the clinical trials legislation, related legal provisions are also changed accordingly as is outlined in Figure 1. These will become applicable as from the date of entry into application of the Clinical Trials Regulation only.
New Provisions GMP for Investigational Medicinal Products
The manufacture and importation of an investigational medicinal product (IMP) requires a manufacturing (or import) authorisation (Article 13 of Directive 2001/20/EC). The holder of a manufacturing (or import) authorisation for IMPs is required to comply with the principles of good manufacturing practices (GMP) for IMPs. The GMP principles for IMPs are currently adopted in Commission Directive 2003/94/EC [6], the ‘GMP Directive’ which applies to both, medicinal products and IMPs. Detailed GMP guidelines for the manufacturing of IMPs are provided in EU GMP Annex 13. [7] In consequence of the revision of the clinical trials provisions the ‘GMP Directive’ had to be revised and will be replaced by two separate legal texts (Figure 1):
- Commission Directive 2017/1572 [8] will apply to medicinal products for human use “whose manufacture or import requires the authorisation referred to in Article 40 of Directive 2001/83/EC”.
- Commission Delegated Regulation (EC) 2017/1569 [9] specifies the principles and guidelines of GMP for IMPs for human use “the manufacture and import of which requires an authorisation as referred to in Article 61(1) of Regulation (EU) 536/2014 and lays down arrangements for inspections of manufacturers in relation to compliance with good manufacturing practice in accordance with Article 63(4) of that Regulation.”
In addition, the current EU GMP Annex 13 (which relates to the GCP Directive 2001/20/EC) was revised to comply with the requirements of the new Clinical Trials Regulation and will be repealed by the ‘Detailed Commission guidelines on good manufacturing practice for medicinal products for human use’. [10]
Specific Provisions Apply for IMPs that are ATMPs
It should be emphasized that IMPs which are at the same time advanced therapy medicinal products (ATMPs) – these are medicinal products based on gene therapies, somatic cell therapies and tissue engineered products – are governed by Regulation 1394/2007. [11] On 22 November 2017, the Commission adopted new guidelines for the good manufacturing practices for ATMPs [12] in order to take into account the unique characteristics of these specific classes of ‘personalized’ medicinal products. The ATMP GMP guideline applies to both, approved ATMPs (ATMPs that have been granted a marketing authorisation) and to ATMPs that are being tested or used as reference in a clinical trial, so called advanced therapy investigational medicinal products. The ‘GMP for ATMPs’ is supplemented to EudraLex Volume 4 (Good Manufacturing Practice (GMP) guidelines) as a new EU GMP Part IV – GMP requirements for Advanced Therapy Medicinal Products (ATMPs) (Figure 1). Figure 1 provides a high-level overview of the current and future provisions that apply for clinical trials in the EU – with focus on those provisions that apply for GMP for IMPs. The boxes highlighted in orange colour indicate the legal framework that applies for clinical trials regulated under the current regimen of Directive 2001/20/EC. The boxes placed in the green field provide an outlook to the new provisions applicable to clinical trials that will be authorised under the Clinical Trials Regulation, once it becomes applicable. The black lines point out the interrelationship between the provisions, red lines indicate where provisions will be repealed.
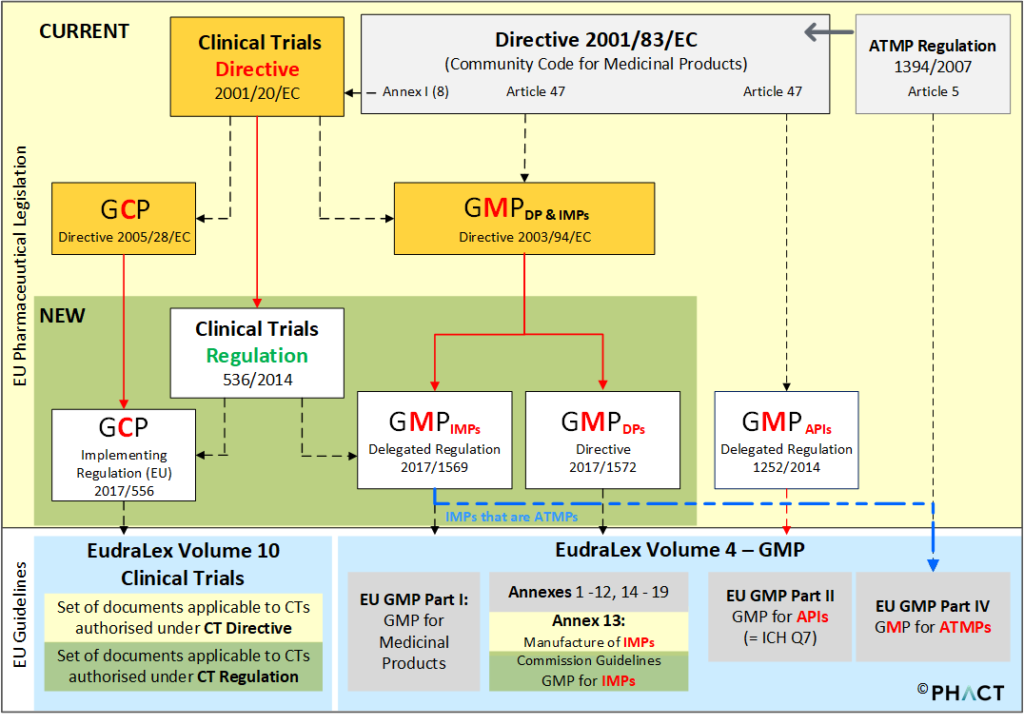
Detailed information about the implementing measures and the status quo of the implementing and delegated acts as are required by the new Clinical Trials Regulation is published on the website of the EU Commission. [13] In order to avoid any confusion, the EU Commission has published two different sets of documents that are applicable to clinical trials authorised under Directive 2001/20/EC or to those that are applicable to clinical trials that will be authorised under Regulation EU No 536/2014, once it becomes applicable. [14] In this respect it should be noted that a 3 years transitional period will start from when the Clinical Trial Regulation becomes applicable. During this transition period, both sets of documents as published on the EU Commissions website will apply.
Abbreviations
| ATMP | Advanced therapy medicinal product |
| CT | Clinical trial |
| DP | Drug product (also referred to as ‘medicinal product’) |
| EC | European Commission |
| EU | European Union |
| GMP | Good manufacturing practice |
| IMP | Investigational medicinal product |
References
- Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC.
- Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use.
- EU Commission’s website: Clinical Trials – Regulation (EU) No 536/2014; https://ec.europa.eu/health/human-use/clinical-trials/regulation_en (accessed July 17, 2019).
- Functional specifications for the EU portal and EU database to be audited. https://www.ema.europa.eu/documents/other/functional-specifications-european-union-eu-portal-eu-database-be-audited_en.pdf (accessed 17 July 2019).
- Website of the European Commission. https://ec.europa.eu/health/human-use/clinical-trials_en (accessed 17 July, 2019).
- Commission Directive 2003/94/EC of 8 October 2003 laying down the principles and guidelines of Good Manufacturing Practice in respect of medicinal products for human use and investigational medicinal products for human use.
- EudraLex The Rules Governing Medicinal Products in the European Union. Volume 4. EU Guidelines to Good Manufacturing Practice. Medicinal Products for Human Use. Annex 13. Investigational Medicinal Products. ENTR/F/2/AM/an D(2010) 3374 as of 03 February 2010. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2009_06_annex13.pdf; accessed July 17, 2019.
- Commission Directive (EU) 2017/1572 of 15 September 2017 supplementing Directive 2001/83/EC of the European Parliament and of the Council as regards the principles and guidelines of good manufacturing practie for medicinal products for human use.
- Commission Delegated Regulation (EU) 2017/1569 of 23 May 2017 supplementing Regulation (EU) No 536/2014 of the European Parliament and of the Council by specifying principles of and guidelines for good manufacturing practice for investigational medicinal products for human use and arrangements for inspections.
- Detailed Commission guidelines on good manufacturing practice for investigational medicinal products for human use, pursuant to the second subparagraph of Article 63(1) of Regulation (EU) No 536/2014. C(2017) 8179 final as of 8 December 2017. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-10/guideline_adopted_1_en_act_part1_v3.pdf (accessed 17 July, 2019).
- Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004.
- EudraLex – The Rules Governing Medicinal Products in the European Union. Volume 4. Good Manufacturing Practice. Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products. Adoption by the European Commission on 22 November 2017.
- Implementation measures by the Commission in the context of Regulation (EU) No 536/2014 – overview and state of play. https://ec.europa.eu/health/sites/health/files//files/clinicaltrials/overview_clinical_trials.pdf (accessed July 17, 2019).
- Official Website of the European Commission. EudraLex – Volume 10 – Clinical trials guidelines. https://ec.europa.eu/health/documents/eudralex/vol-10_en, accessed 17 July 2019.
Legal provisions referenced in Figure 1 that are not mentioned in the text
- Commission Delegated Regulation (EU) No 1252/2014 of 28 May 2014 supplementing Directive 2001/83/EC of the European Parliament and of the Council with regard to principles and guidelines of good manufacturing practice for active substances for medicinal products for human use. https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1562782270992&uri=CELEX:32014R1252 (accessed 10 July 2019).
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (Consolidated version). https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf (accessed 17 July 2019).
- Commission Directive 2005/28/EC of 8 April 2005 laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such product. https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1562774130954&uri=CELEX:32005L0028 (accessed 17 July 2019). (“GCP Directive”)
- Commission Implementing Regulation (EU) 2017/556 of 24 March 2017 on the detailed arrangements for the good clinical practice inspection procedures pursuant to Regulation (EU) No 536/2014 of the European Parliament and of the Council. https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1562774033514&uri=CELEX:32017R0556 (accessed 17 July 2019).